
max planck institut
informatik
informatik

A test prediction has been run on the Glur2 receptor (PDB ID: 1m5d) with a target rotamer density of 2. That means that IRECS assigns on average two conformations two each side chain. The less constrained and the more flexible a side chain is, the more conformations it receives.
Both pictures visualize some part of the predicted protein. The crystal structure is colored in blue as a reference, the predicted side-chain conformations are colored according to their effective energy. Occupancy values for ambiguous atom positions are calculated from this energy with the Boltzmann law. Pictures were made using PyMOL
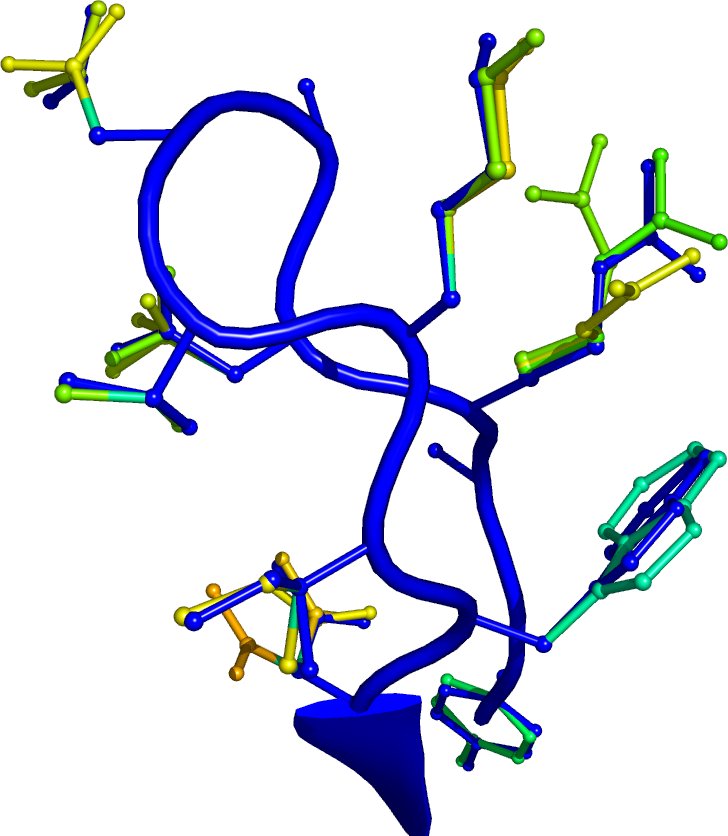
|
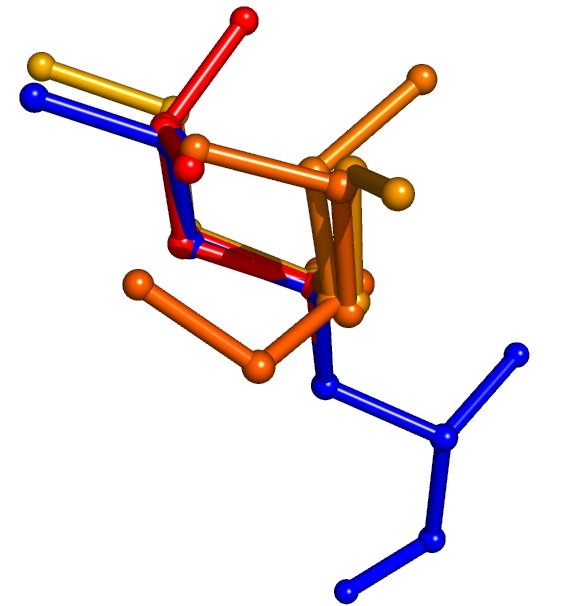
|
|
Tyrosine 61 to Glycine 73. The sequence of the displayed segment is YGARDADTKIWNG. |
Lysine 258. IRECS assigns seven conformations to it, the correct conformation has the lowest effective energy and the highest occupancy |
Below you see an excerpt of the IRECS output in PDB format. The temperature field holds the computed effective energy of the whole rotamer
After installation, run the program with the help option '-h'. You should then see a screen similar to this:
Please note that the target sequence must have the same number of characters as the template has residues. Since IRECS does not model the target backbone, every gap character in the target sequence will be interpreted as a mutation to glycine.
You can see a test run of IRECS below. The protein 1M5D (PDB ID) can be found in the IRECS package.